


Abstract:
Caspase-1 triggers programmed necrosis called pyroptosis by gasdermin D (GSDMD) cleavage. However, GSDMD-deficient cells are still susceptible to caspase-1-mediated cell death. Researchers at Kanazawa University etc in Japan discovered that caspase-1 proteolytically activates Bid and initiates apoptosis in GSDMD-deficient cells. Furthermore, cortical neurons and mast cells, exhibiting little GSDMD expression, undergo apoptosis after appropriate stimulation in a caspase-1- and Bid-dependent manner. This study clarifies molecular mechanism and biological roles of caspase-1-induced apoptosis in GSDMD-low/null cells.
[Background]
Most unnecessary or dangerous cells such as virus-infected cells commit suicide by activating an in-built suicide program. This type of cell death is referred to as “programmed cell death”. Until about year 2000, it was considered that apoptosis*1) is almost the sole mode of programmed cell death at least in mammals. However, in recent years, various modes of “non-apoptotic” programmed cell death such as pyroptosis*2) and necroptosis have been defined based on the molecular mechanisms and morphological characteristics. In the process of apoptosis, the group of proteases called caspases, about 10 species of which are found in humans, function as suicide enzymes. Caspase-1*3), the first caspase found in mammals, has been reported to induce apoptosis in neurons in animal models of neurological disorders such as cerebral infarction, Alzheimer’s disease and amyotrophic lateral sclerosis (ALS). On the other hand, subsequent studies revealed that in macrophages*4) infected by bacteria, caspase-1 induces necrosis*5)-like and inflammatory programmed cell death, which was thereafter named pyroptosis. Thus, caspase-1 has been reported to induce apoptosis or pyroptosis depending on the biological context (Figure 1). The reason for this apparent discrepancy has not been clarified.
Recently, it was revealed that pyroptosis is induced by proteolysis of a protein called gasdermin D (GSDMD)*6) by caspase-1. Nonetheless, macrophages whose GSDMD gene was destroyed exhibited cell death upon activation of caspase-1, although more slowly than wild-type macrophages. Therefore, it seemed that there should be a mechanism of caspase-1-mediated cell death independent of GSDMD. However, the form and molecular mechanism of such cell death remained unknown.
[Results]
The present research team led by scientists of Kanazawa University together with scientists from Hokkaido University, the University of Tokyo and National Institute of Genetics, Japan, investigated the caspase-1-mediated programmed cell death of macrophages. First, they found that GMDSD-deficient macrophages underwent apoptosis when they were infected with Salmonella, although wild-type macrophages underwent pyroptosis under the same conditions. Since caspase-1-deficient macrophages exhibited little cell death under the same conditions, it was clear that both modes of programmed cell death, pyroptosis and apoptosis, were mediated by caspase-1.
Next, the team investigated caspase family members required for caspase-1-mediated apoptosis in GSDMD-deficient cells. As a result, caspase-3 and caspase-9 were found to be involved; caspase-3 is known to be necessary for induction of a broad spectrum of apoptosis, while caspase-9 is activated in response to cytochrome c release from mitochondria. Then, Bid*7), a pro-apoptotic cytoplasmic protein, emerged as a candidate for a mediator of caspase-1-induced apoptosis because of the following reasons. Bid is a well-known substrate of caspase-8, and its cleaved product, tBid induces cytochrome c release from mitochondria and subsequent caspase-9 activation. Furthermore, it has been reported that Bid can also be cleaved by caspase-1. Thus, the team generated GSDMD/Bid double deficient cells; on activation of caspase-1, it was found that apoptosis was suppressed in these cells compared to the GSDMD-deficient cells. Furthermore, GSDMD/Bid double deficient macrophages were found to be more resistant to Salmonella infection than GSDMD deficient macrophages. These results indicated that in GSDMD-deficient cells, caspase-1 activated Bid instead, which induced apoptosis.
In previous studies, caspase-1 was reported to be involved in the apoptosis of spinal cord neurons in mouse models of familial amyotrophic lateral sclerosis and of cortical neurons after oxygen/glucose deprivation. It was also demonstrated that such apoptosis was accompanied by Bid cleavage (activation) in a caspase-1-dependent manner. However, it was not clear whether Bid was necessary for the programmed death of those neurons or whether cortical and spinal cord neurons expressed GSDMD. Thus, the team investigated the expression of GSDMD mRNA and protein in the cerebral cortex and the spinal cord, and found that little GSDMD is expressed in these tissues. Moreover, cultured cortical neurons from caspase-1-deficient mice and those from Bid-deficient mice under oxygen/glucose deprivation were less susceptible to apoptosis in comparison with wild-type neurons. In addition, by consulting BioGPS (http://biogps.org/, a public database of protein expression), mast cells, a kind of immune cell, were found to express caspase-1 and Bid, but little GSDMD. Accordingly, when mast cells were stimulated by nigericin, a bacterial toxin, which is known to induce pyroptosis in macrophages, mast cells exhibited caspase-1- and Bid-dependent apoptosis.
The results of the present study revealed that apoptosis was induced even with neurons and mast cells that express little GSDMD by activation of Bid through caspase-1 action.
[Future prospects]
Caspase-1 is expressed in various cell-types and tissues besides macrophages and neurons, and has been suggested to be involved in myocardial infarction and renal disorders in addition to neurodegenerative diseases. It is necessary to elucidate whether in these cases, caspase-1-dependent cell death is GSDMD-dependent pyroptosis or Bid-dependent apoptosis or even due to other programmed cell death mechanisms.
In the future, by developing selective inhibitors for individual modes of programmed cell death in specific cells based on their underlying mechanisms, improvements may be expected in the prevention and treatment of diseases caused by programmed cell death.
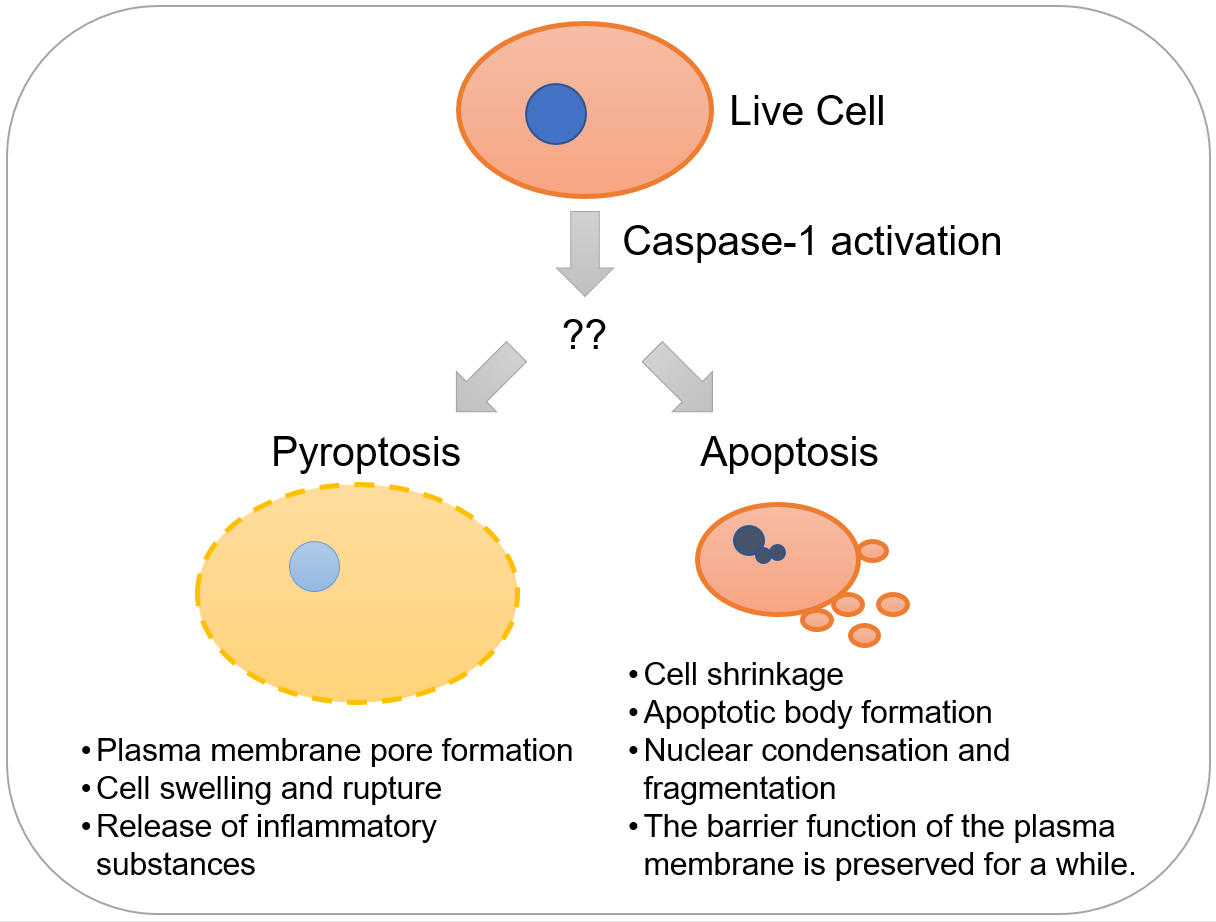
Figure 1. Characteristics of two modes of programmed cell death induced by caspase-1 activation.
Recently, it has been demonstrated that activated caspase-1 induces pyroptosis, inflammatory and necrosis-like programmed cell death, although it was previously thought to induce apoptosis. It was not known why caspase-1 induces two different modes of programmed cell death with distinctive characteristics depending on the biological context.
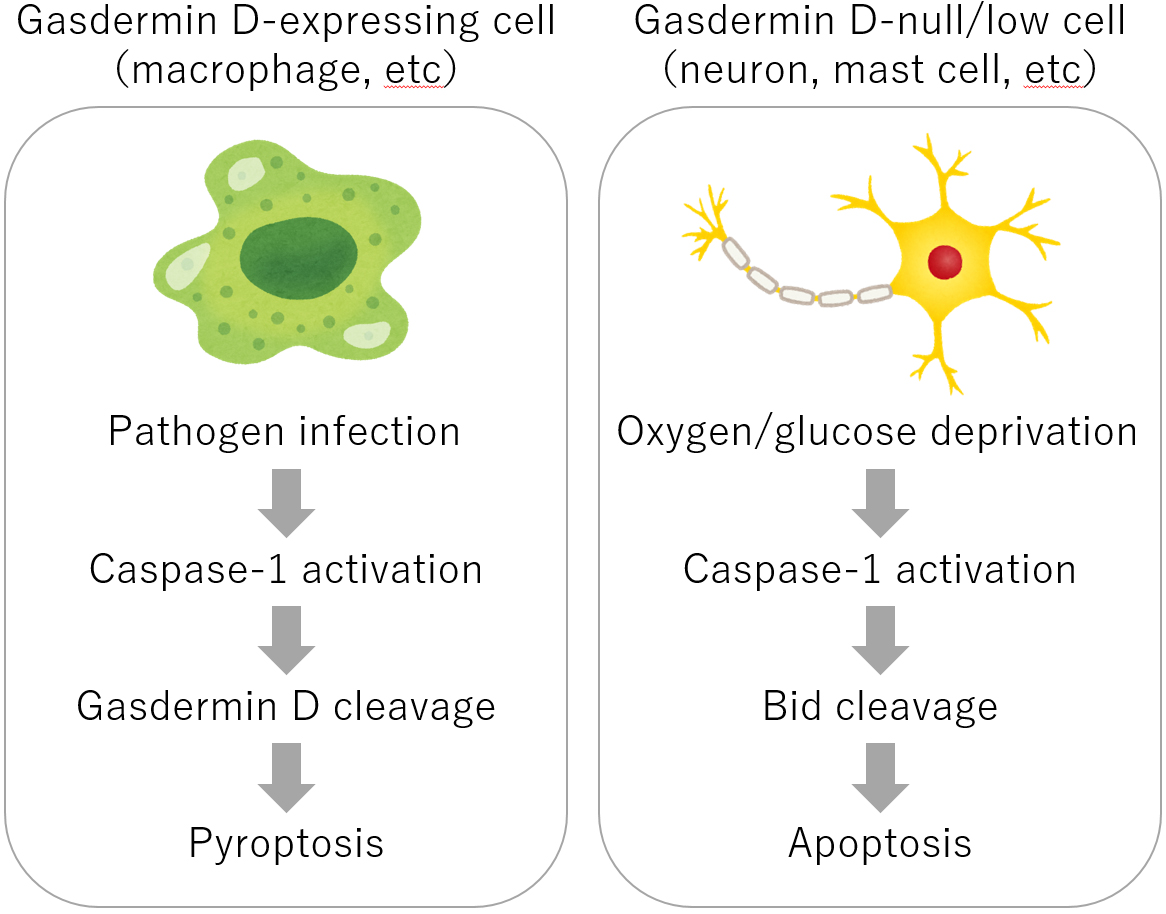
Figure 2. Mechanism of the two modes of programmed cell death induced by caspase-1.
Caspase-1 induces pyroptosis in those cells that express gasdermin D such as macrophages. On the other hand, in the case of those cells like neurons that do not express gasdermin D, it induces apoptosis though Bid activation.
[Glossary]
*1) Apoptosis
A mode of programmed cell death, induced by activation of caspase. Apoptosis is involved in a variety of phenomena such as development, immunity and suppression of cancer. Apoptotic cells exhibit characteristic morphological changes such as cell shrinkage, formation of small extracellular vesicles called apoptotic bodies from the cell membrane, condensation and fragmentation of the nucleus, whereas the barrier function of the cell membrane is maintained for some time. In the meantime, phosphatidyl serine, a phospholipid usually present in the inner leaflet of the plasma membrane, is exposed on the cell surface, and functions as a factor inducing phagocytosis. Because of this, an apoptotic cell is rapidly phagocytosed by an adjacent cell or a macrophage. This process limits the release of intracellular substances that induce inflammation. Therefore, it is generally considered that apoptosis does not induce inflammation, unless a large number of cells simultaneously undergo apoptosis, exceeding the processing capacity of macrophages.
*2) Pyroptosis
Pyroptosis is a mode of inflammatory programmed cell death induced by activation of caspase-1, 4 or 5. Pyroptotic cells rupture rapidly, and release intracellular inflammatory substances. Because of this, a strong inflammation is induced in surrounding tissue. Pyroptosis functions as protection of the body against bacterial infection but it can lead to diseases such as septic shock.
*3) Caspase-1
Caspase-1 is an intracellular protease. When activated in response to various stresses such as pathogen infection to a cell, it induces programmed cell death. It also acts on the immature (inactive) forms of interleukin-1β (IL-1β) or IL-18 to convert them into their mature (active) forms, which then induce inflammation.
*4) Macrophage
A type of leucocyte with a high capability for phagocytosis. Macrophages phagocytose and eliminate bacteria, but some highly pathogenic bacteria may act as parasites and propagate in macrophages.
*5) Necrosis
Necrosis is a mode of cell death, characterized by rapid cell membrane disruption. Leakage of intracellular inflammatory substances causes inflammation. It was thought that necrosis was non-programmed cell death, primarily due to extrinsic stress. Recently, however, various modes of programmed cell death like pyroptosis and necroptosis that show morphological characteristics similar to necrosis have been discovered.
*6) Gasdermin D
A cytoplasmic protein substrate of caspase-1. Amino-terminal fragments of GSDMD cleaved by caspase-1 assemble with each other, forming pores in the cell membrane. Through the pores, water and other molecules flow into the cell, causing cell rupture.
*7) Bid
Bid is a cytoplasmic protein, which can be proteolytically activated by caspase-8. Upon activation, it causes release of cytochrome c from mitochondria and induces apoptosis. It was known that caspase-1 also cleaves Bid; however, the significance of this was unknown.
Article
Caspase-1 initiates apoptosis in the absence of gasdermin D
Journal: Nature Communications
Authors: Kohsuke TSUCHIYA1, Shinsuke NAKAJIMA1, Shoko HOSOJIMA1, Dinh Thi NGUYEN1, Tsuyoshi HATTORI1, Thuong Manh LE1, Osamu HORI1, Mamunur Rashid MAHIB1, Yoshifumi YAMAGUCHI2, Masayuki MIURA3, Takeshi KINOSHITA1, Hiroko KUSHIYAMA1, Mayumi SAKURARI1, Toshihiko SHIROISHI4 and Takashi SUDA1
1. Kanazawa University, 2. Hokkaido University, 3. The University of Tokyo, 4. National Institute of Genetics
DOI: 10.1038/s41467-019-09753-2
Funder
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas (KAKENHI Grant Number 26110002) and by Grant-in-Aid for Challenging Exploratory Research (KAKENHI Grant Number 15K15078) from the Japan Society for the Promotion of Science (to T. Suda). This work was also supported by the Japan Society for the Promotion of Science (KAKENHI Grant Number 26460523) and the Institute for Frontier Science Initiative (InFiniti), Kanazawa University (to K.T.).