


Abstract:
Tumor tissues include cancer stem-like cells (CSCs) that are responsible for recurrence and drug resistance. The metabolic mechanisms in CSCs are largely unknown and currently there are no therapies targeting them. We show that methylenetetrahydrofolate dehydrogenase 2 (MTHFD2)-mediated purine synthetic metabolism is essential for the maintenance of CSCs. Targeted therapy against MTHFD2 would be a plausible solution to the unresolved problems of recurrence and drug resistance to improve the prognosis of cancer patients.
An increase in the incidence and mortality rate of cancer has been noted worldwide, especially for lung cancer. Many patients with lung adenocarcinoma harbor mutations in epidermal growth factor receptor (EGFR), and initially show a good response to molecular target drugs, such as gefitinib, which inhibits the EGFR tyrosine kinase. However, recurrence inevitably occurs within a few years because of acquired drug resistance, leading to poor prognosis. Despite the development of new generations of molecular targeted drugs, recurrence and acquired drug resistance remain problematic.
Emerging evidence suggests that cancer stem-cells (CSCs) are responsible for tumor recurrence and drug resistance. Tumor tissues comprise heterogeneous cell types including CSCs and their differentiated progeny, as observed in normal tissues derived from tissue-specific stem cells. Most cancer therapies, such as conventional chemotherapy, radiotherapy, and molecular target drugs, target rapidly proliferating differentiated cancer cells, but not CSCs. It is thought that cancer cells with stem-like properties continue to survive after treatment, and a fraction of these cells exhibit the ability to re-divide after years, causing recurrence. Thus, targeted therapies against CSCs and their daughter cells are important for tumor eradication; however, this is still an area of unmet medical need. To establish such therapies, it is important to investigate the molecular mechanisms that determine how CSCs are maintained in cancer tissues. Although it is believed that various metabolic pathways are activated in cancer cells, the specific metabolic mechanisms in CSCs remain largely unclear.
One-carbon (1C) metabolism incorporates carbons as building blocks of purine and pyrimidine that are used for DNA replication and RNA transcription, and thus plays an important role in cell proliferation (Figure). In the mitochondria, methylenetetrahydrofolate dehydrogenase (MTHFD2), an enzyme involved in 1C metabolism, is strongly expressed in cancer cells, while it is scarcely expressed in normal cells. MTHFD2 is required for cancer cell proliferation. Active MTHFD2 consumes 5-aminoimidazole carboxamide ribonucleotide (AICAR), an intermediate of the purine synthesis pathway, to produce IMP, a purine nucleotide.
We discovered that it is essential to continuously consume AICAR and reduce its concentration in cancer cells to low levels for the maintenance of CSCs. Active MTHFD2 is critical for this mechanism. Therefore, MTHFD2-mediated mitochondrial 1C metabolism appears critical for the survival of CSCs and resistance to drugs including gefitinib through consumption of AICAR, leading to depletion of the intracellular pool of AICAR. Because CSCs are dependent on MTHFD2, therapies targeting MTHFD2 may eradicate tumors and prevent recurrence.
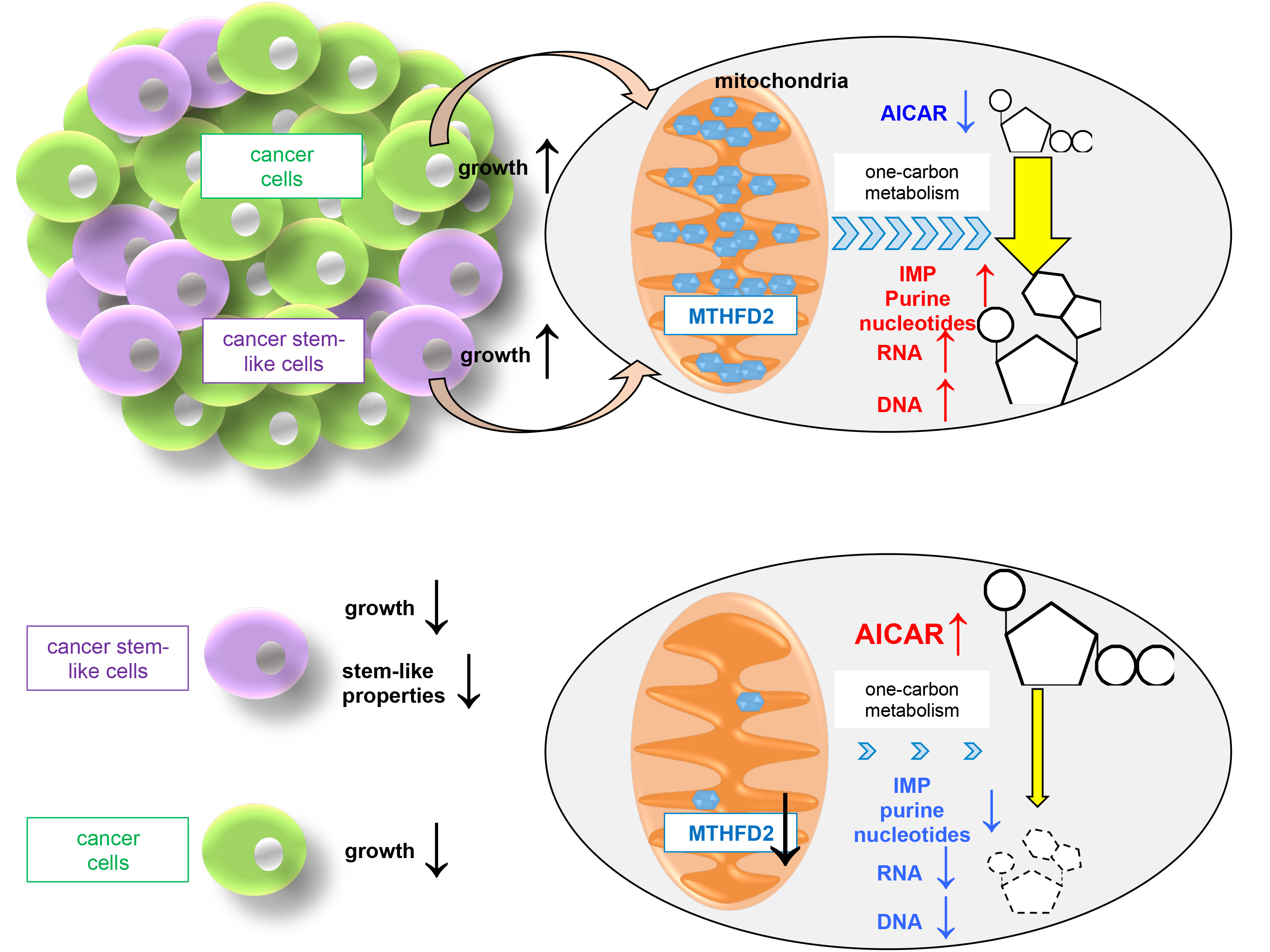
Figure.
Upper panel: MTHFD2 is abundantly expressed in the mitochondria in cancer cells. Active MTHFD2-mediated one-carbon metabolism consumes AICAR and produces IMP and purine nucleotides for cancer cell proliferation. Reduced intracellular pool of AICAR is essential for maintenance of cancer stem-like cells and their growth.
Lower panel: When MTHFD2 expression is inhibited, cancer cell proliferation is inhibited due to reduced production of IMP and purine nucleotides and cancer stem-like cells are reduced due to increase levels of AICAR.
Article
Cancer stem-like properties and gefitinib resistance are dependent on purine synthetic metabolism mediated by the mitochondrial enzyme MTHFD2
Journal: Oncogene
Authors: Tatsunori Nishimura, Asuka Nakata, Xiaoxi Chen, Kurumi Nishi, Makiko Meguro-Horike, Soichiro Sasaki, Kenji Kita, Shin-ichi Horike, Kaori Saitoh, Keiko Kato, Kaori Igarashi, Takahiko Murayama, Susumu Kohno, Chiaki Takahashi, Naofumi Mukaida, Seiji Yano, Tomoyoshi Soga, Arinobu Tojo, Noriko Gotoh
DOI: 10.1038/s41388-018-0589-1
Funders
This work was supported in part by an Extramural Collaborative Research Grant from the Cancer Research Institute, Kanazawa University, Hokkoku Gan-Kikin, a Grant-in-Aid for Scientific Research on Innovative Areas from MEXT (22130009), a Grant-in-Aid for Scientific Research from JSPS (15H04294, 17K19587), and a research grant from AMED Project for Development of Innovative Research on Cancer Therapeutics. This work was supported in part by a Grant-in-Aid for Scientific Research for Young Scientists from JSPS (17K15021).