


Abstract:
Researchers at Kanazawa University report in Proceedings of the National Academy of Sciences of the United States of America (PNAS) that a particular signaling pathway in breast-cancer tumors causes cancer cells to divide symmetrically, expanding the tumor. Inhibiting the pathway by drugs could become a strategy for eliminating the cancer cells.
In breast cancer, one of the most common cancers in women, tumors contain a small amount of so-called cancer stem-like cells (CSCs). Being able to eliminate breast-cancer stem-like cells in a targeted way is essential for developing successful therapies — conventional treatments, such as chemotherapy or radiotherapy followed by drug intake, do not target CSCs. A better understanding of the processes generating CSCs in breast-cancer tumors is needed. Noriko Gotoh from Kanazawa University and colleagues have now uncovered a signaling pathway directly related to the proliferation of CSCs in breast cancer.
A property of stem cells is that they can self-renew and differentiate. Two types of stem-cell division can occur: symmetric or asymmetric (Fig. 1). In the former, two self-renewing stem cells are generated; in the latter, only one (and a differentiated cell). It is now believed that malignant CSCs have a higher tendency to divide symmetrically, thus increasing the number of CSCs. Gotoh and colleagues looked at how the microenvironment of CSCs, called the CSC niche, causes and sustains an increased rate of symmetric division.
The researchers started from the observation that a particular gene encoding a type of cytokine known as Semaphorin 3 (Sema3) was one of the most-expressed genes in the CSC niche (Fig. 2). (Cytokines are small proteins that, when released, affect the behavior of cells around them.) The production of Sema3 activates another protein, called MICAL3, the expression levels of which were also found to be high in the CSC niche.
Via a series of experiments in vitro, Gotoh and colleagues were able to confirm the critical roles of Sema3 and MICAL3 in breast cancer tumor development. Specifically, MICAL3 was shown to be required for tumor sphere formation (tumorigenicity is associated with spherical cell shapes). The scientists showed that Sema3-stimulated MICAL3 triggered a whole sequence of biomolecular interactions (a signaling pathway), ultimately resulting in induced symmetric division of CSCs, and hence their proliferation, in breast cancer (Fig. 3).
Having established this important pathway is highly relevant for developing treatments for breast cancer, because, quoting Gotoh and colleagues, “by inhibition of MICAL3 … or knockdown of each component in the signaling pathway, the symmetric cell division may be inhibited, leading to a reduction of breast-cancer stem-like cells.”
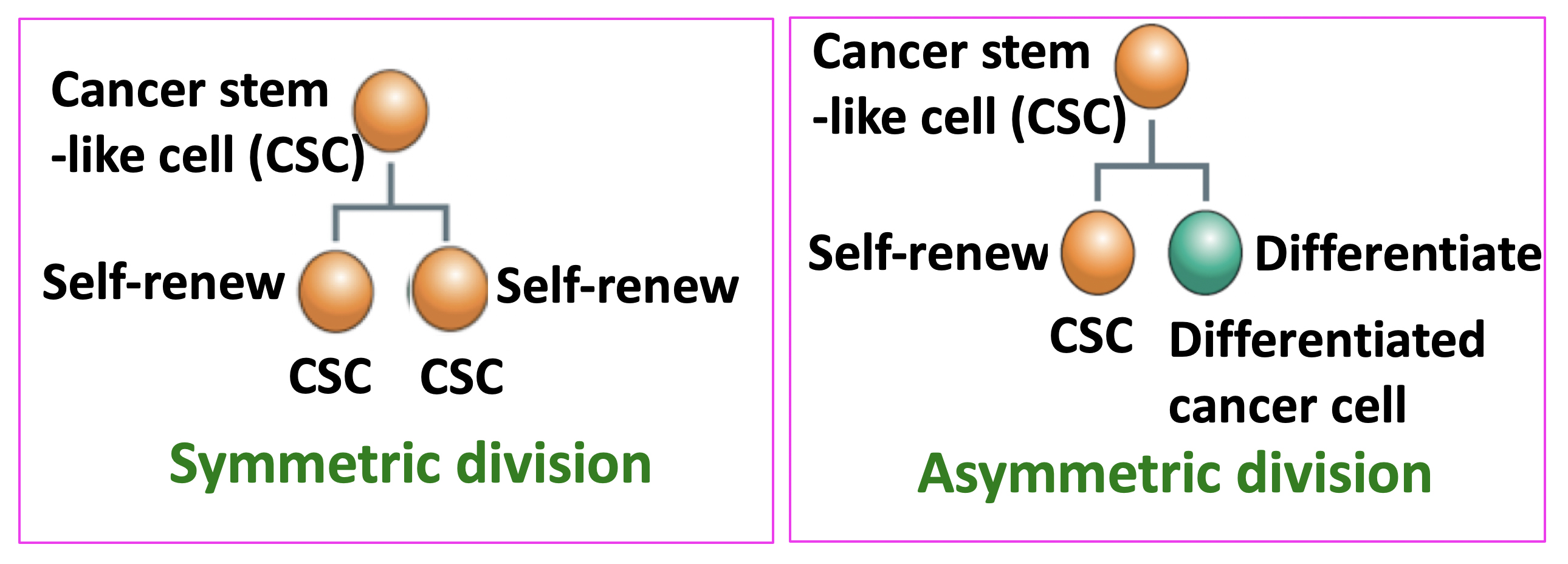
Figure 1.
Cancer stem-like cells (CSCs) undergo two types of cell division. CSCs can self-renew and differentiate. Two types of cell division can occur: symmetric or asymmetric. In the former, two self-renewing CSCs are generated; in the latter, only one (and a differentiated cell).
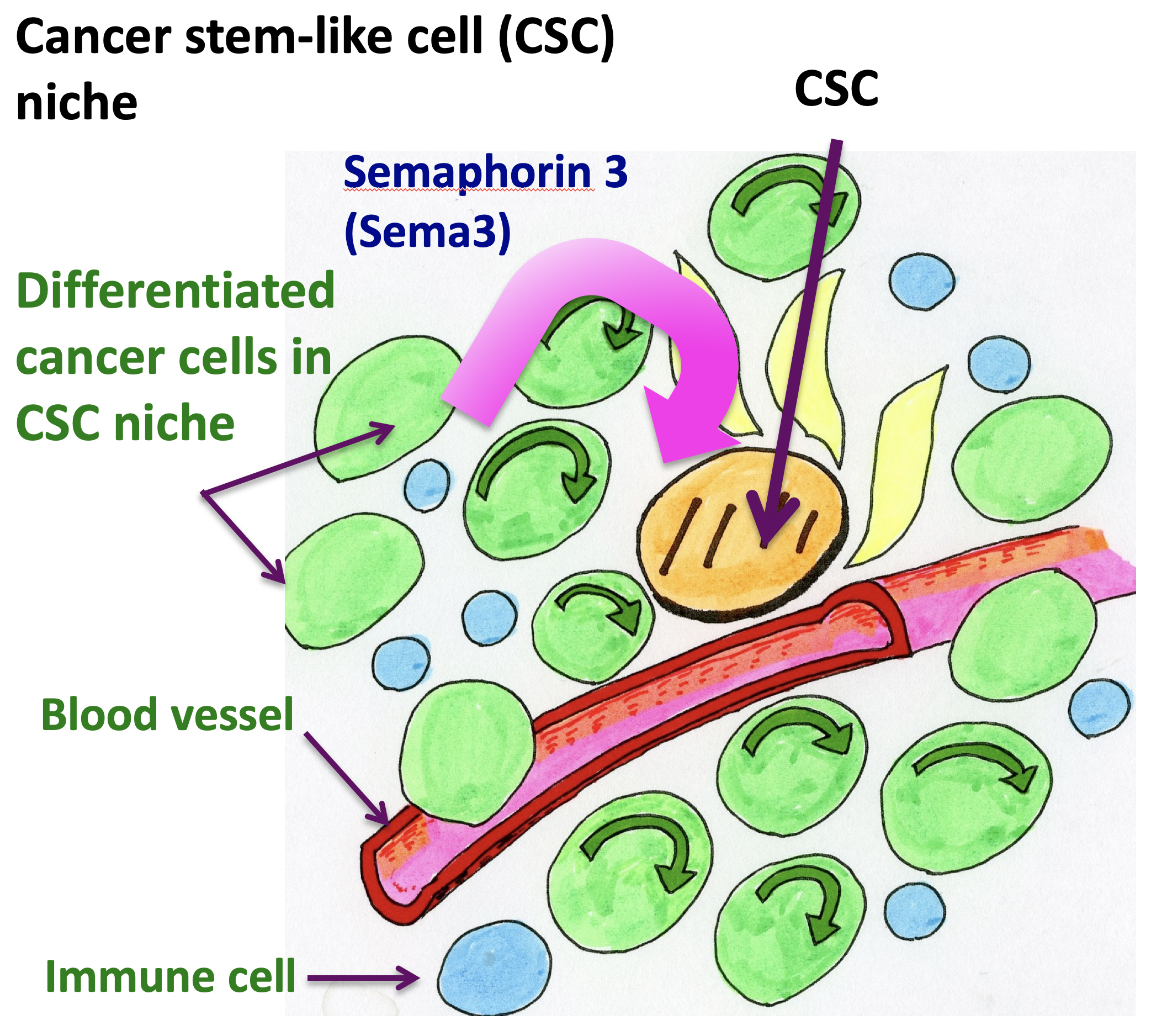
Figure 2.
Semaphorin 3 (Sema3) produced by differentiated cancer cells in CSC niche affects CSCs. CSC niche contains various kinds of cells, including CSCs, differentiated cancer cells. endothelial cells in blood vessels, and immune cells. Sema3 is mainly produced by differentiated cancer cells in CSC niche.
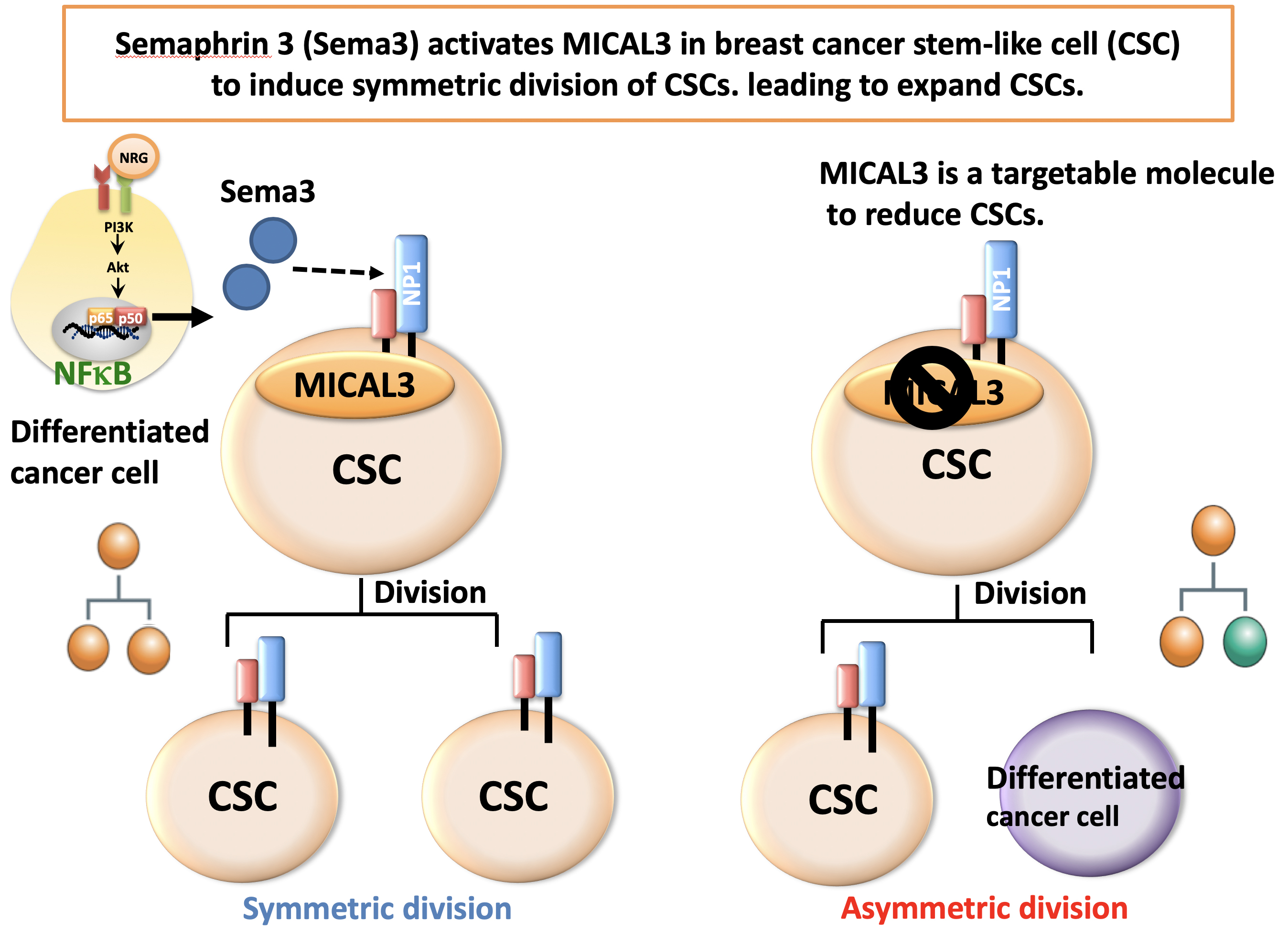
Figure 3.
Sema3 activates MICAL3 in breast CSCs and induces symmetric division of CSCs, leading to expand CSCs. MICAL3 is a targetable molecule to reduce CSCs.
Article
Semaphorin signaling via MICAL3 induces symmetric cell division to expand breast cancer stem-like cells
Journal: Proceedings of National Academy of Science of the United States of America (PNAS)
Authors: Authors: Kana Tominaga, Hiroshi Minato, Takahiko Murayama, Asako Sasahara, Tatsunori Nishimura, Etsuko Kiyokawa, Hajime Kanauchi, Seiichiro Shimizu, Ayaka Sato, Kotoe Nishioka, Ei-ichi Tsuji, Masao Yano, Toshihisa Ogawa, Hideshi Ishii, Masaki Mori, Koichi Akashi, Koji Okamoto, Masahiko Tanabe, Kei-ichiro Tada, Arinobu Tojo, and Noriko Gotoh
DOI: 10.1073/pnas.1806851116
Funders
This work was supported in part by an Extramural Collaborative Research Grant from the Cancer Research Institute, Kanazawa University, Hokkoku Gan-Kikin, a Grant-in-Aid for Scientific Research on Innovative Areas from MEXT (22130009), a Grant-in-Aid for Scientific Research from JSPS (15H04294, 17K19587 and 18H02679), and a research grant from AMED Project for Development of Innovative Research on Cancer Therapeutics, Project for Cancer Research and Therapeutic Evolution and Practical Research for Innovative Cancer Control to N. Gotoh.